La enfermedad de Wilson es una enfermedad rara que me resultó
bastante llamativa ya que consiste en la acumulación de cobre en los tejidos, motivo por el cual he decidido tratar este tema.
¿Qué es?
La enfermedad de Wilson o degeneración hepatolenticular es
una enfermedad hereditaria autosómica recesiva.
Se caracteriza por una alteración en el transporte y
eliminación del cobre que proviene de la dieta, lo que provoca con el paso del
tiempo el depósito de este metal en órganos.
Principalmente afecta al hígado, al encéfalo y a la córnea del ojo, con
los consiguientes fallos en los mismos.
Su síntoma principal es la acumulación de cobre en los
tejidos, manifestada por síntomas neurológicos (dificultad de coordinación,
temblores), también catarata y enfermedades hepáticas (hepatitis, cirrosis, insuficiencias).
La enfermedad afecta por igual a hombres y a mujeres y se ha descrito en todas
las razas.
La enfermedad de Wilson es considerada una enfermedad rara,
su frecuencia en casi todas las poblaciones estudiadas es de uno por cada
40.000 habitantes. La frecuencia de las personas portadoras de la mutación es
cercana a un 1%.
¿Qué sucede con el metabolismo del cobre?
Se estima que en el cuerpo hay unos 50-100 mg de cobre que
son necesarios para llevar a cabo diversas funciones del metabolismo. Y
mediante la comida ingerimos todos los días unos 2-5 mg de este oligoelemento.
En los pacientes con enfermedad de Wilson, el metabolismo
intermediario del cobre está alterado debido a una mutación en el gen ATP7B.
Este gen codifica una proteína, llamada de la misma forma, que es necesaria
para la eliminación del cobre por la bilis y para que el cobre pase a la
ceruloplasmina, que es la proteína que transporta el cobre por la sangre. Al no
existir una correcta eliminación del cobre a través de la bilis, hay una
tendencia a la acumulación excesiva de este metal, lo que causa lesiones
hepáticas que pueden comenzar incluso a partir de los tres años de vida.
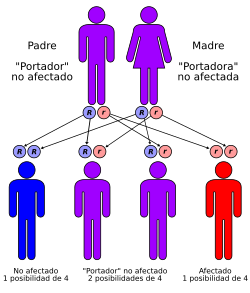
¿Cómo se hereda?
La enfermedad de Wilson se hereda de forma autosómica
recesiva, es decir, que para que aparezca la enfermedad hay que heredar la
mutación en el gen ATP7B tanto del padre como de la madre. Las personas que
heredan la mutación de uno de sus padres pero no del otro son portadores de la
mutación, pero no desarrollan la enfermedad.
La mutación en el gen ATP7B no es siempre la misma, sino que
se han descrito en la literatura médica más de 350 mutaciones posibles en este
gen.
¿Cómo y cuándo se manifiesta la Enfermedad de Wilson?
La enfermedad de Wilson afecta principalmente al hígado,
siendo una de sus manifestaciones más tempranas la inflamación de este órgano,
lo que se conoce en términos médicos como hepatitis. Cuando esto ocurre se
produce una liberación al torrente sanguíneo de las enzimas hepáticas,
conocidas como las transaminasas, y de la bilirrubina, sustancia responsable
del color amarillento que adquiere la piel y mucosas de estos pacientes
(ictericia). La hepatitis tiende a curarse espontáneamente, pero se puede
repetir en varias ocasiones y el paciente puede acabar por desarrollar un
proceso inflamatorio crónico del hígado, que hace que el tejido normal se
destruya y sea sustituido por tejido fibroso, originando lo que se conoce como
cirrosis hepática. Cuando esto ocurre el hígado es incapaz de funcionar
adecuadamente.
El otro tipo de manifestación clínica de la enfermedad de
Wilson ocurre a nivel neuro-psiquiátrico, y se caracteriza por alteraciones del
movimiento, como descoordinación y temblores, dificultad para el habla y
problemas para tragar los alimentos. También puede presentarse pérdida de
memoria, disminución del rendimiento intelectual, dolores de cabeza,
alteraciones del comportamiento, manifestaciones psicóticas o neuróticas,
síntomas de demencia y crisis convulsivas.
Igualmente es muy frecuente la afectación el globo ocular. La
parte más afectada por el depósito inadecuado del cobre es la córnea, en su
diámetro más externo, produciéndose un fenómeno conocido como anillos de
Kayser-Fleischer. Este anillo se produce por el depósito de cobre inicialmente
en los polos superiores e inferiores de la córnea y con posterioridad rodeando
el iris. Este anillo se puede detectar en una exploración oftalmológica, lo que
puede ayudar al diagnóstico de enfermedad de Wilson. Por otro lado se puede
producir endurecimiento del cristalino y aumento de la opacidad del mismo, lo
que se denomina cataratas.
Diagnóstico
Lo primordial para poder diagnosticar la enfermedad de Wilson
es la sospecha clínica, basada en los signos y síntomas del paciente y en las
alteraciones en las pruebas de la función hepática. Ante una sospecha clínica
se pueden realizar los siguientes estudios:
-Cuantificación de cobre en la orina de 24 horas: valores superiores a 100 microgramos /día son habituales en estos pacientes (los valores para una persona sana oscilan entre 20 y 50 microgramos/día).
-Niveles de cobre libre en suero: los valores de cobre libre en las personas que sufren esta enfermedad son mayores de 25 microgramos/dl.
 -Niveles de ceruloplasmina en suero: están disminuidos en esta enfermedad, generalmente por debajo de 20 mg/dl.
-Niveles de ceruloplasmina en suero: están disminuidos en esta enfermedad, generalmente por debajo de 20 mg/dl.
-Presencia de los anillos de Kayser-Fleischer en el ojo: esta alteración se detecta habitualmente por un oftalmólogo utilizando una lámpara especial llamada lámpara de hendidura.
-Test genético para detectar en el ADN del paciente la
mutación del gen, para lo que se usan técnicas de secuenciación genética.
También es importante el cribado de familiares de primer grado menores de 40
años.
-Estudios de imagen: si el paciente presenta síntomas
neuro-psiquiátricos se suele solicitar
una RMN (resonancia magnética nuclear) del cerebro, lo que permite detectar
depósitos de cobre y atrofia cerebral.
Tratamiento
La enfermedad de Wilson no se cura de forma definitiva, sólo
pueden aplicarse tratamientos de por vida dirigidos a controlar el depósito de
cobre en el organismo.
El tratamiento usado con mayor frecuencia es una dieta pobre en cobre, aunque no existe una indicación para mantener una dieta estricta baja en cobre dada la capacidad de los fármacos actuales. Sin embargo, conviene avisar al paciente de cuáles son para no abusar de ellos. Algunos alimentos muy ricos en cobre son los frutos secos, los mariscos, los champiñones, el chocolate, la gelatina y la soja.
En segundo lugar, desde el punto de vista terapéutico, los
medicamentos disponibles actualmente están dirigidos a disminuir los depósitos
de cobre, mediante un proceso químico llamado quelación, es decir, los fármacos
se unen al cobre de forma irreversible y facilitan su eliminación vía renal o
intestinal. Los más comunes son la penicilamina (cada vez más en desuso por sus
efectos adversos a nivel neurológico), la trientina y el acetato de cinc.
Las últimas recomendaciones apuntan a iniciar el tratamiento
con acetato de cinc lo más pronto posible, incluso en pacientes portadores homocigotos de la mutación que aún no hayan mostrado síntomas. El acetato de
cinc, además, es el tratamiento de elección en embarazadas y en niños.
En tercer lugar, si el paciente lo requiere, es decir si
tiene insuficiencia hepática, se puede llevar a cabo un trasplante hepático.
Investigadores españoles desarrollan una terapia génica que cura la enfermedad de Wilson
Os dejo el link de este tan interesante artículo sobre unos investigadores españoles que desarrollan una terapia génica que cura la enfermedad de Wilson.
http://www.abc.es/salud/enfermedades/abci-investigadores-espanoles-desarrollan-terapia-genica-cura-enfermedad-wilson-201602160615_noticia.html
A continuación, os dejo un vídeo que he encontrado que explica esta enfermedad:
Fotos:
https://es.wikipedia.org/wiki/Enfermedad_de_Wilson#/media/File:Autorecessive-es.svg
https://s-media-cache-ak0.pinimg.com/564x/75/15/a1/7515a16cf44df4ca0c00e163ee6e5125.jpg
http://www.webconsultas.com/sites/default/files/styles/cabecera_categoria/public/temas/enfermedad-wilson_1.jpg?itok=hRwcBd6t
Información:
http://eusalud.uninet.edu/misapuntes/index.php/Enfermedad_de_Wilson
https://es.wikipedia.org/wiki/Enfermedad_de_Wilson
http://www.webconsultas.com/categoria/salud-al-dia/enfermedad-de-wilson
http://www.abc.es/salud/enfermedades/abci-investigadores-espanoles-desarrollan-terapia-genica-cura-enfermedad-wilson-201602160615_noticia.html
Vídeo:
https://www.youtube.com/watch?v=gbZtw9QyFik